Barriers to Entry and Barriers to Validation
Rethinking IP for biomarkers
In the final section of my recent keynote address for Cancer Research UK‘s 2019 Symposium on Oesophageal Cancer, I departed from my usual rants on technical rigor in science to discuss the political barriers that keep us from effectively translating and scaling new diagnostics in cancer. In the talk, I discuss a number of failures of incentives in both academia and industry that fall into this category. Many thanks to Profs. Fitzgerald and Lovat for the opportunity to not just speak but to be provocative in my address to the audience. The slides are available here if you’re interested (start on slide 36 for the content of this post).
Biomarker development, costs, and intellectual property
Diagnostics development is expensive. Traditionally, validity and reimbursement for diagnostics rest on a triad of analytical validity (“can you accurately measure the physical thing you claim to measure?”), clinical validity (“does the thing you measure actually correlate with the disease state you care about?”), and clinical utility (“even if you can measure disease, does it change health decisions or outcomes?”). The first is the most straightforward of the lot. The second is difficult: it’s the biological validation problem that I discussed in the first two-thirds of the talk and have covered previously here and elsewhere. The last is also challenging as it requires actually testing the interaction of your diagnostic with the care system.
At the end of a biomarker discovery project, we could assume that we have decent coverage of analytical validity, and a hypothesis for clinical validity that needs to be tested in a larger study. This larger validity study is likely to be very expensive because of the hundreds to thousands of patients who will need to be tested: sample recruitment, sample acquisition, and sample processing are all generally pretty pricey. Note that this looks similar to trials in therapeutics: once you clear preclinical studies (analogous to the “discovery” study), you go into human clinical trials which are even more expensive.
In our capitalist system, these expensive trials will not be conducted if the private-sector funder does not think they’ll be able to make a positive return on the capital invested. The big challenge here is that a validation study isn’t meant to discover anything new; it just shows that something known works. In a vacuum, this makes these studies economically unfeasible: you’ll spend a bunch of money proving that drug / diagnostic X works, and then someone else can use your data to go off and sell X without having to invest in that study themselves (a “second-mover advantage” or “free rider problem”). This is the foundation for intellectual property protection (primarily patents and trade secrets) in pharmaceutical development: because we as a society believe that we gain from having new drugs, we incentivize the upfront R&D costs to bring new drugs to market by providing a period of exclusivity. However, this period is time-limited, as we also believe that it is important to share this knowledge broadly and thereby make a therapy available cheaply once those initial costs have been paid down.
However, court decisions in the last ten years have significantly weakened the intellectual property regime around clinical diagnostics and biomarkers. (Note: I am not a lawyer, and this post is not a legal opinion; it is only my understanding of the landscape.) In the wake of the Prometheus, Myriad, Alice, and Athena cases, it is unclear what subject matter in biomarkers and diagnostics is in fact patentable, as many biomarkers are correlations between physiological states and disease states that could be argued to be “laws of nature”. While unique laboratory processes are probably patentable, because there are often multiple ways to measure the same underlying signal, protecting one method may not be enough to protect the investment in a large clinical validity study.
The digging of data moats
It is hotly argued whether this change in IP protection and incentives has negatively affected investment in novel diagnostics and personalized medicine. It is undoubtedly true that companies have had to respond to this climate in order to justify their investments, and one strategy that has cropped up more and more is the “data moat”. This strategy is inspired by what we’ve seen in the non-bio tech world: companies like Google and Facebook are able to maintain dominating positions even in the presence of open technology stacks because they have a lead in data collected that is nearly impossible to surmount; even if you copied their code, you could copy neither their accumulated data nor their inflow of data, and so they will continue to outperform.
The idea goes as follows: if you can’t patent a biomarker signature itself, maybe you can do the following instead:
- Build up a large data set of clinical samples on which to do discovery.
- Use an automated algorithm to discover a biomarker and build a test.
- Patent the laboratory and algorithmic processing.
- Validate and market the test as a black box so that copying of the test is difficult or impractical.
- Continuously optimize the test based on new samples to maintain a lead.
This approach or one similar to it motivates a number of “data plays” in clinical diagnostics, ranging from Myriad Genetics’ strategy of keeping its variant (re)classification database private, to various companies’ efforts at using machine learning for pathology and diagnosis.
While this “data moat” strategy can partially address the challenges around IP and barriers to entry for a second-mover, I believe it creates a number of broader concerns. First, regulators and the public will have reasonable concerns regarding steps 2 and 4: how can you validate a black box, and why should anyone believe that such a test is valid? Beyond that, the entire goal of the strategy is to block competition; if successful barriers for second movers may arise that are too high, where no reasonable competitor can emerge and a winner-take-all market appears. This has already been hugely controversial in consumer technology, and would be very different from how other medical technology is treated. While we grant patent rights on drugs to incentivize innovation, but time-limit those rights so that society can benefit from generic competition and price reduction in the long run (market failures in biosimilars aside). This seems desirable in the diagnostic space as well, but data moats admit no mechanism for genericization.
How can we realign the incentives for diagnostics validation?
Although the case law makes sense as the law is written, it is leading to the emergence of a competitive strategy that is suboptimal for society: we would like to incentivize development of new medical technology, but at the same time we have a vested interest in this kind of care becoming broadly available and inexpensive. It’s therefore imperative to think about models for diagnostics development and funding that might close this gap.
One option would be to scale up public, consortium, or charity funding in the vein of previous large projects like the PLCO trial, the 1000 Genomes Project, etc. While this might fix the funding challenge for the validation trial proper, it would not be a complete solution. While this would address clinical validity, it still would not address clinical utility, reimbursement, etc., and so kicks the problem one step down the line. Obviously, it also would be a political challenge to secure additional funds to do these studies (or to cut funding from other worthwhile projects).
A second option is to redesign IP protection for diagnostics. While the existing uncertainty of protection is incentivizing potentially harmful competitive behavior and concentration, forcing diagnostics into the existing patent regime may also be inappropriate. First, modern diagnostics are economically distinct from pharmaceuticals. Whereas both have large upfront R&D costs, pharmaceuticals typically have much lower unit production costs (biologics excluded) than typical diagnostics COGS, and have a longer path through discovery and the clinic, making breakeven timelines different. Furthermore, although diagnostics validation studies are not designed to discover new biology, it is likely especially with the modern use of high-content assays like NGS and shotgun mass spectrometry that the data sets generated in validation studies could be used for interesting new discovery projects. Consequently, alternatives to the conventional long-duration patent regime may be worthwhile. One interesting model may be a time-embargoed release model, in which a company that funds and executes a successful validation study receives a period of market exclusivity — but at the end of this period, not only must give up that exclusivity, but must also make public the data from the study. Such a model would both incentivize industry to spend its own funds towards research, but also benefit downstream research with new data and the public with the possibility of generic competition down the line. Similar models have seen success in the genomics world, in which a number of pharmaceutical companies (including Regeneron, GSK, AbbVie, and others) have funded sequencing of the UK Biobank cohort in exchange for a period of exclusive access to the sequencing data.
How do we make these changes happen?
At the beginning of the talk, I asked the audience what they thought was the biggest challenge blocking better cancer diagnostics: basic science, translational science, engineering, or politics. As might be expected from an audience of mostly academic scientists and clinicians, most people thought the problems were scientific in nature:
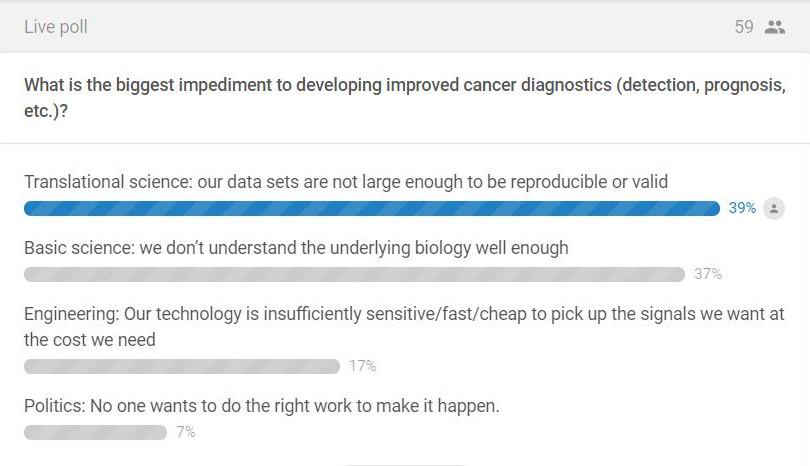
It was gratifying to see answers change by the end of the talk and to see the audience agree with me that political challenges are actually a huge barrier:

This still leaves the question of how to act on these issues. While we could leave this all up to government action, there are actions that can be taken at lower levels including individual companies and labs. For example, I’ve been a strong advocate at my previous employers to share our data as much as possible (open access papers with strong data supplements to enable future research and reanalysis). Establishing norms like these is an important part of fulfilling industry’s contract with society to support future research and advance health. At the same time, it is important to balance this against a recognition that return on investment is a cornerstone of how our society has chosen to structure innovation, and that companies cannot be expected to act as charities if we want to incentivize that innovation. If anyone has clever ideas on how to solve this knotty problem, I’d love to hear about them on Twitter.